About
Small RNAs are major post-transcriptional regulators of gene expression in bacteria. A major challenge has been to decipher the global map of small RNA-target interactions in the bacterial cell. To this end we developed the RIL-seq methodology (RNA Interaction by Ligation and sequencing), which identifies RNA pairs co-bound to the protein Hfq (a chaperon that mediates their interaction). Application of RIL-seq to Escherichia coli K-12 MG1655 grown to different growth phases or under different growth conditions has revealed thousands of small RNA-target interactions, defining a complex post-transcriptional regulatory network that rewires under growth different conditions. To handle the massive amounts of data produced by RIL-seq, we developed RILseqDB - a database that enables querying the revealed interacting RNAs. The database enables search of three types:
- RNA search - Enables viewing all the data available for a RNA of interest, which currently include its interactome and summary of its interactions in the different experiments.
- Interaction search - Enables viewing specific interactions by querying the interacting RNAs. Interactors can be defined by name or by their genomic annotation (e.g. CDS, 3'UTR). Data regarding the interaction is provided as well as visualization of the mapped reads in a genome browser format.
- Shared interactor search - Enables finding shared RNA interactors of several RNAs (specified by the user).
Contact us at RILseqDB.admin@ekmd.huji.ac.il.
RIL-seq Methodology
RIL-seq is an experimental-computational methodology for global identification of small RNA-target pairs in bacteria. The experimental part of RIL-seq takes advantage of the mutual binding of the sRNA and its target to the Hfq chaperone, where their base-pairing takes place. The main steps of RIL-seq involve in-vivo protein-RNA cross-linking, cell lysis, coimmunoprecipitation of Hfq and bound RNAs, RNA ligation, RNA isolation, paired-end sequencing, and mapping of the sequenced fragments to genome of the organism under study. Each sequenced fragment was represented by two sequences at its ends, which were mapped individually to the genome. This mapping revealed two types of fragments: 1) 'single' fragments, where the "end sequences" are mapped to the same genomic location, and thus have most likely originated from a single transcript. 2) 'chimeric' fragments, where the "end sequences" are mapped to two distinct genomic locations, supporting a putative interaction between RNAs derived from these regions.
Experimental Workflow
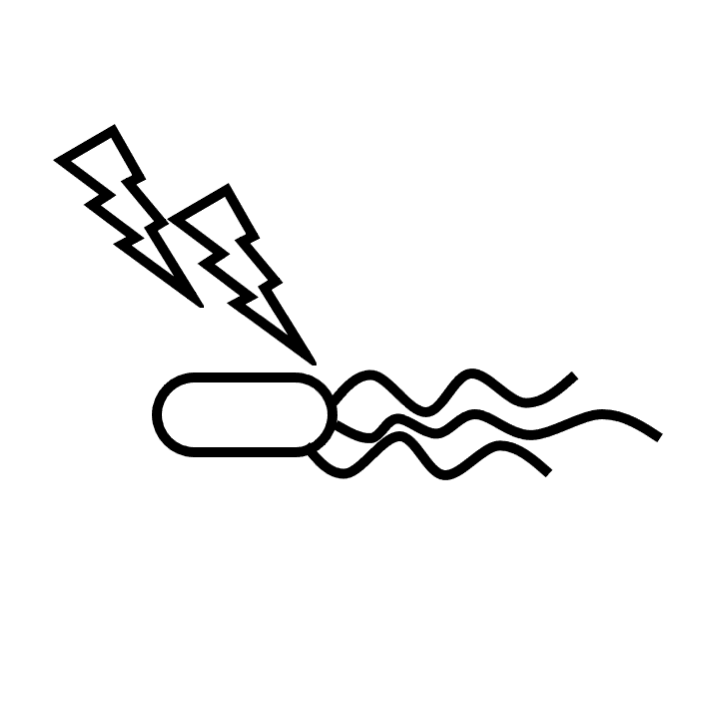
STEP 1: Protein-RNA in vivo cross-linking by UV irradiation
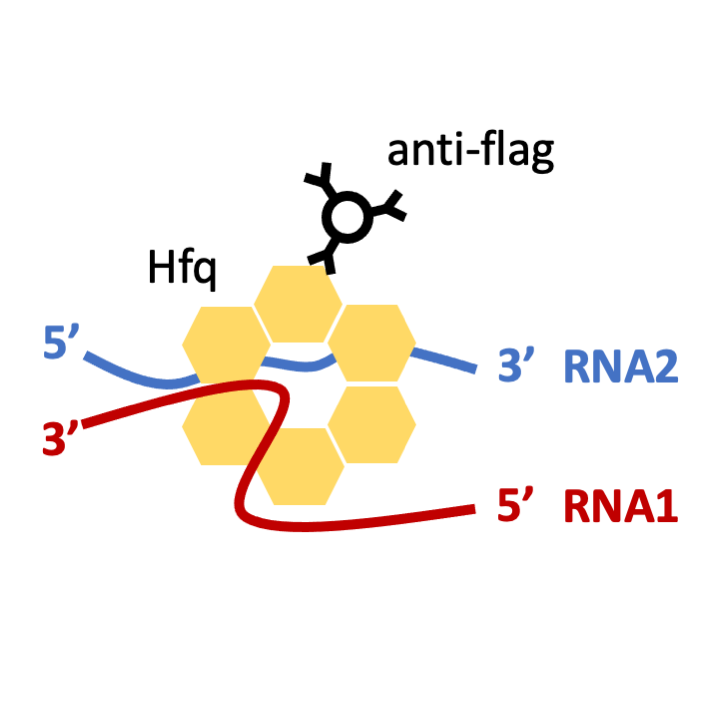
STEP 2: Cell lysis and Hfq-Flag Co-IP with anti-flag antibody
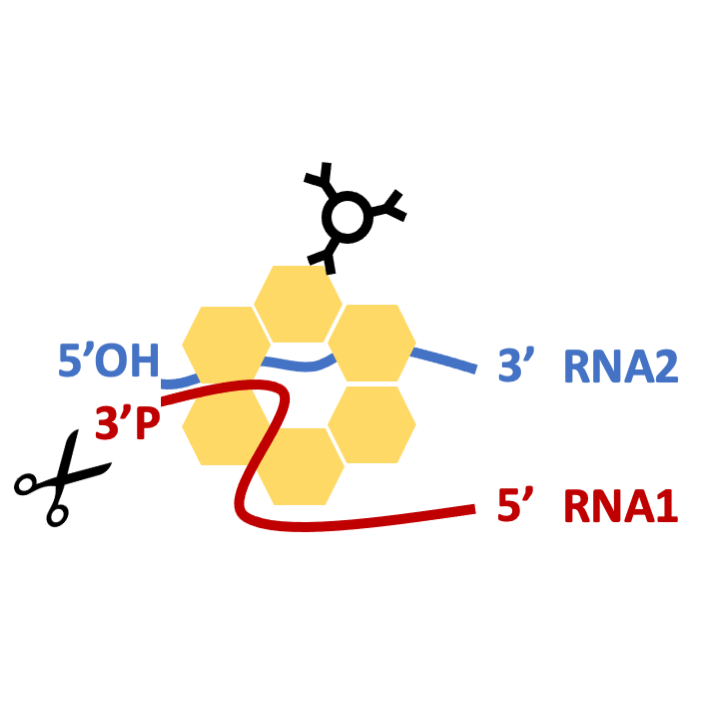
STEP 3: A/T RNases digestion
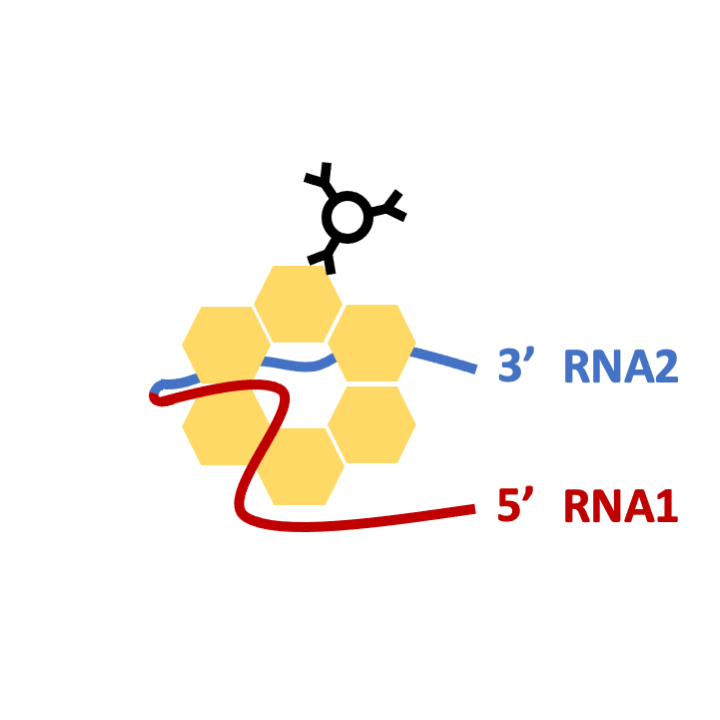
STEP 4: T4 PNK treatment and RNA1-RNA2 ligation
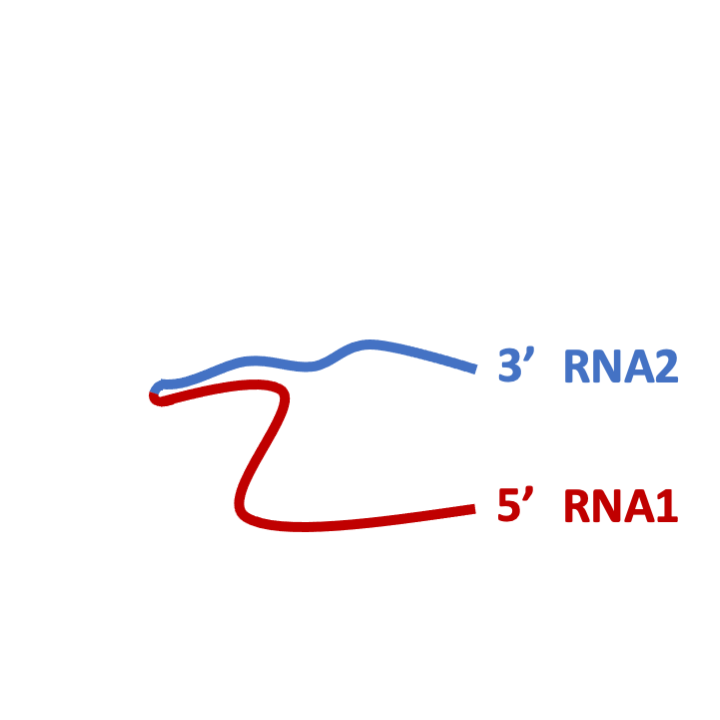
STEP 5: Proteinase K digestion and RNA isolation
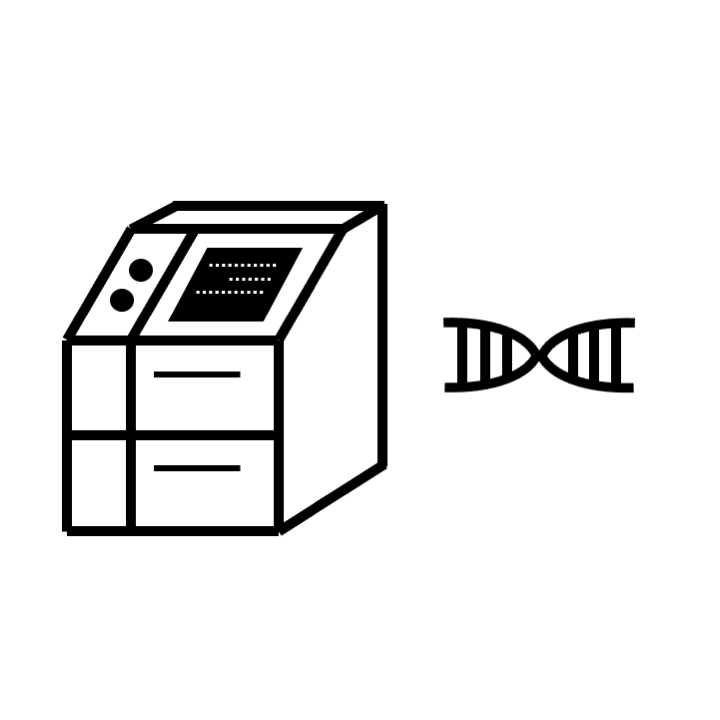
STEP 6: Library construction and Illumina sequencing (paired-end)
Computational Pipeline
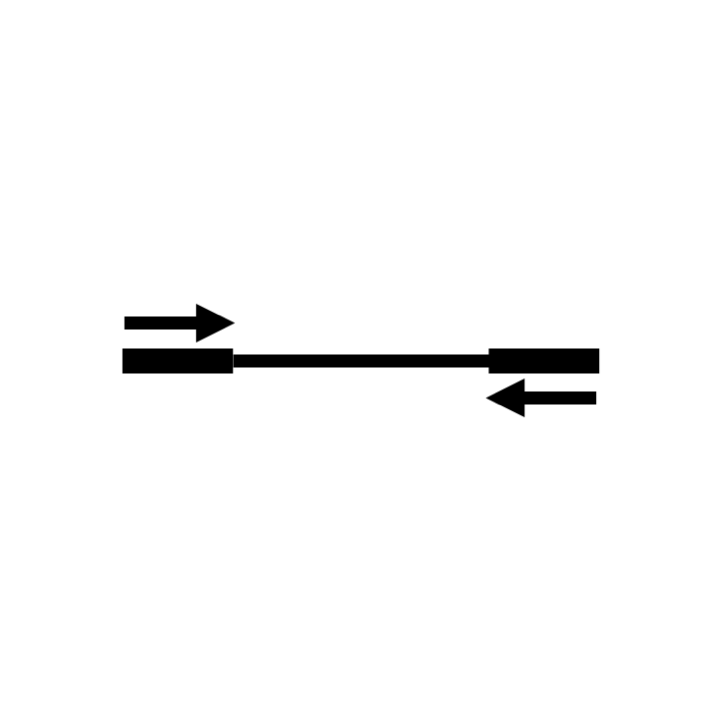
STEP 7: Mapping ends of sequenced fragments
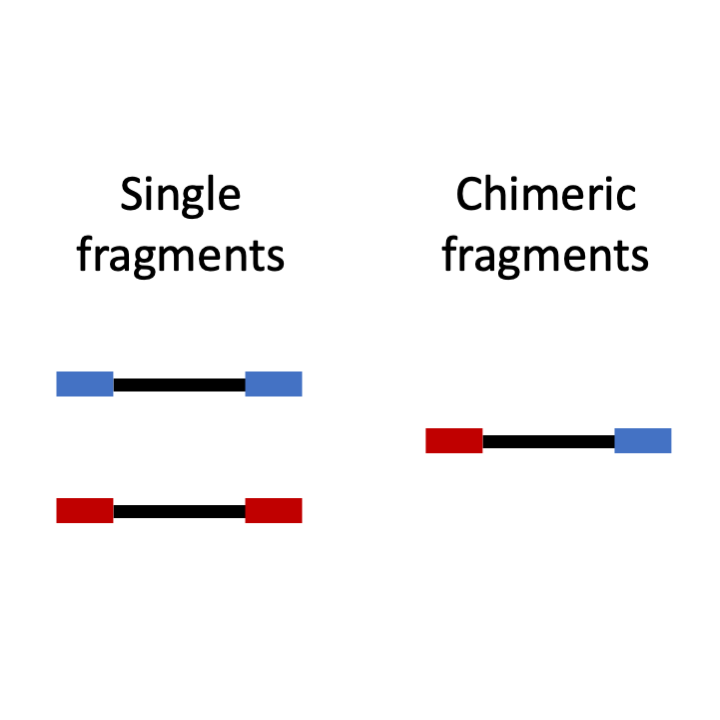
STEP 8: Classifying "single" and "chimeric" fragments
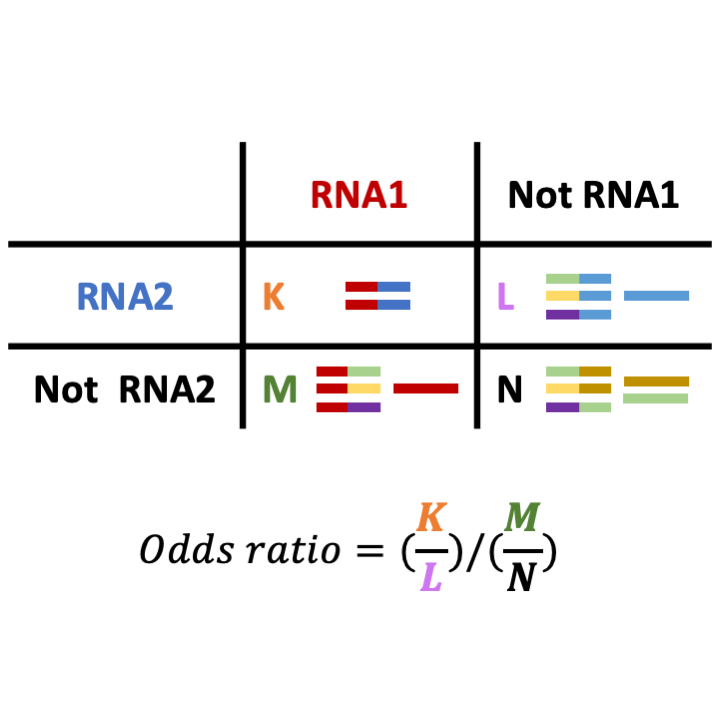
STEP 9: Finding over-represented pairs of regions in chimeric fragments by applying one-sided Fisher's exact test
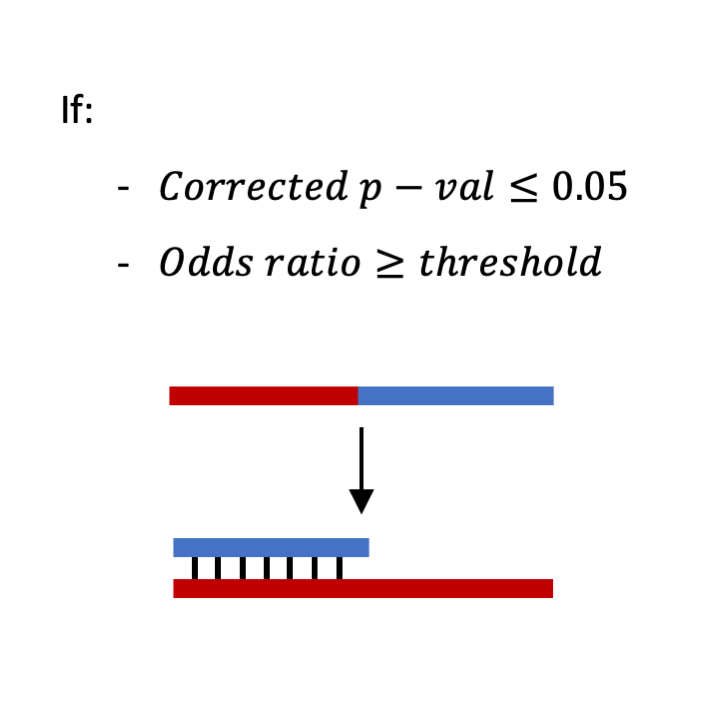
STEP 10: Determining statistically significant chimeras representing RNA-RNA interaction
Data
The current database includes data sets of Escherichia coli K-12 MG1655 and of enteropathogenic E. coli (EPEC).
Contributing Projects
The following projects contributed to this project:
Grants
This study was supported by:
The European Research Council Advanced Grants #322920 and #833598.
The Israel Science Foundation (grants 1411/13 and 876/17).
Israeli Centers of Research Excellence (grants 1796/12 and 41/1).